BeaconGENEsis 1.0 User Guide
BeaconGENEsis is a new Java application to assist in the design of molecular beacons. It creates beacon candidates using an algorithm designed to take into account general design rules and then outputs all of the candidates to a textarea. The user then can use these in the mfold DNA folding program and, using the data from the server, select the top beacon candidates.
After installation, simply click on the icon to start the program. Shown below is a screen shot of the program.
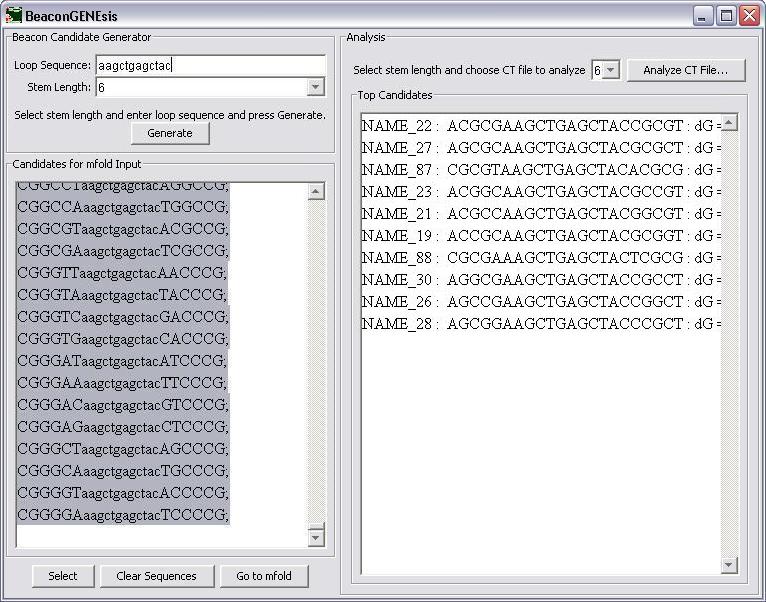
First the loop sequence is entered into the appropriate textbox.
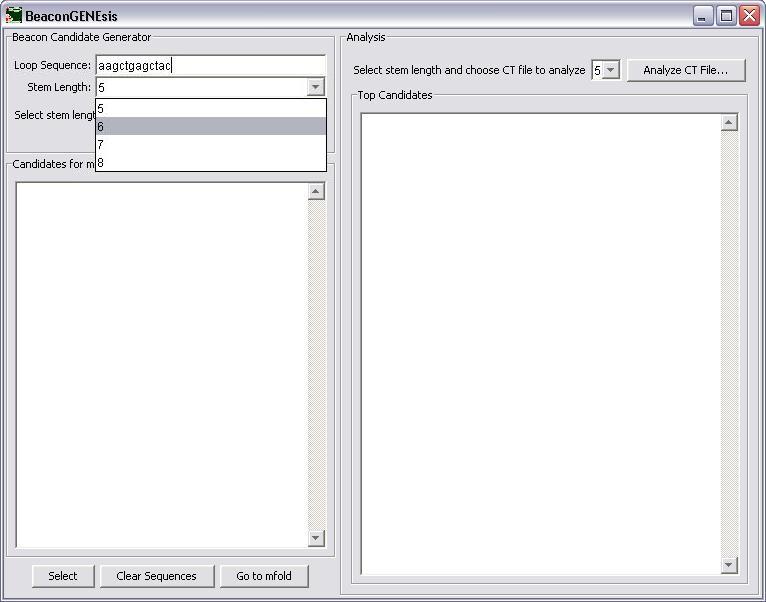
The stem length is then chosen next by selecting from the choices in the combo box.
After the stem length is selected, the Generate button is pressed to generate the candidate sequences for input into mfold.
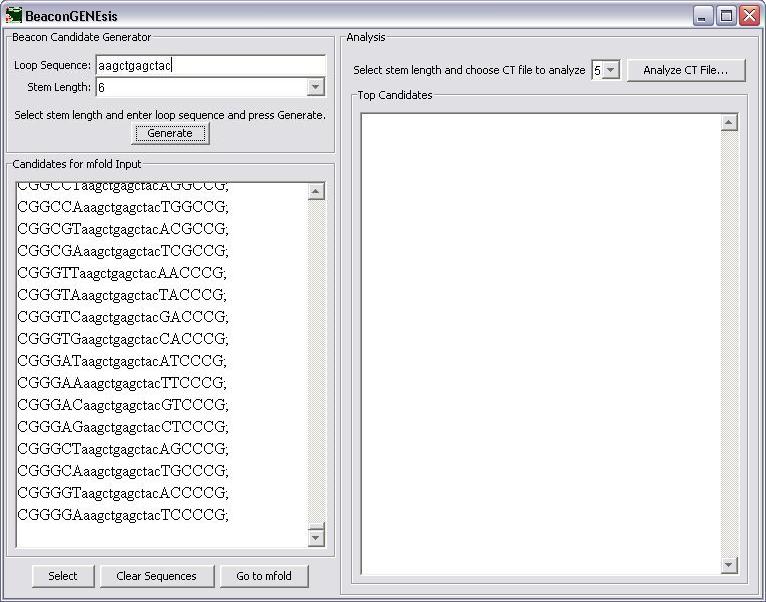
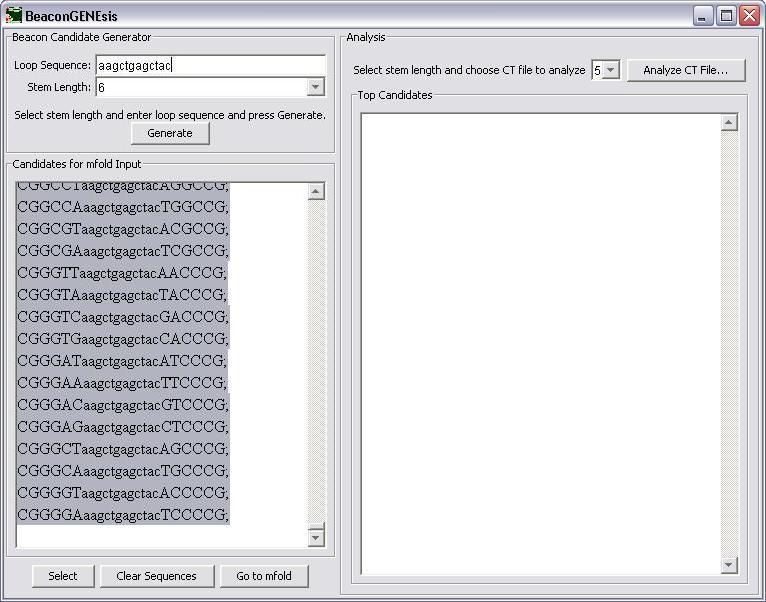
If you would like to change the loop sequence or the stem length and generate different beacons, press the "Clear Sequences" button and then generate the new sequences. If these sequences are correct, press the "Select" button to copy the them and then press "Go to mfold" to go to the mfold server site.
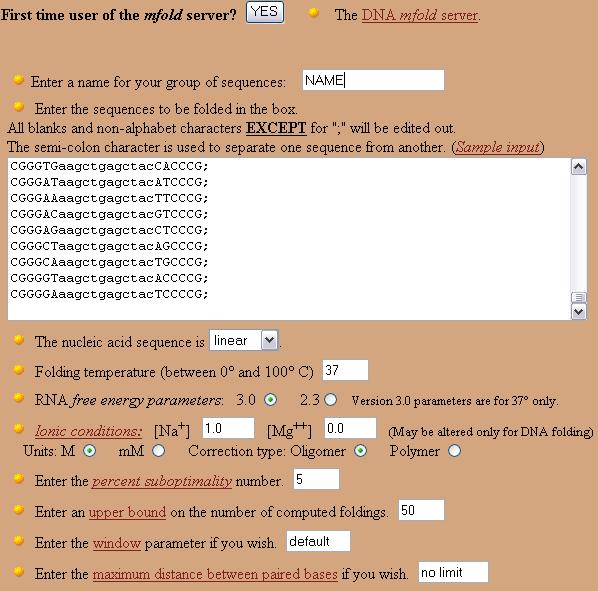
At the mfold site, paste the sequences into the sequence text area and type the name of the sequence in the appropriate text box.
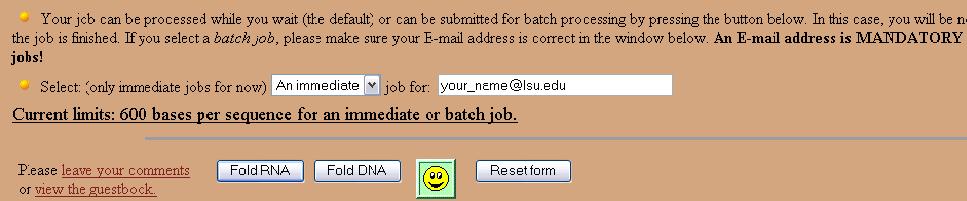
Next, enter an email address and click the "Fold DNA" button.
After the folding is complete, the page shown below will be displayed. Click on "ct file (all foldings concatenated)" to download the CT file.
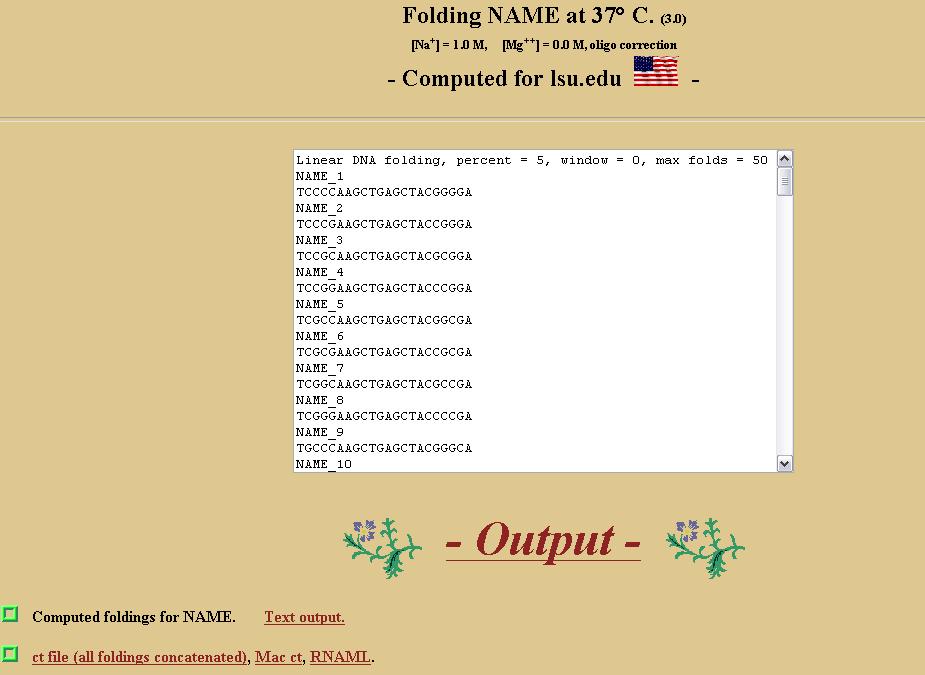
Next, select the stem length of the CT file that you wish to analyze from the choices in the combo box and press "Analyze CT File..." to analyze the file.
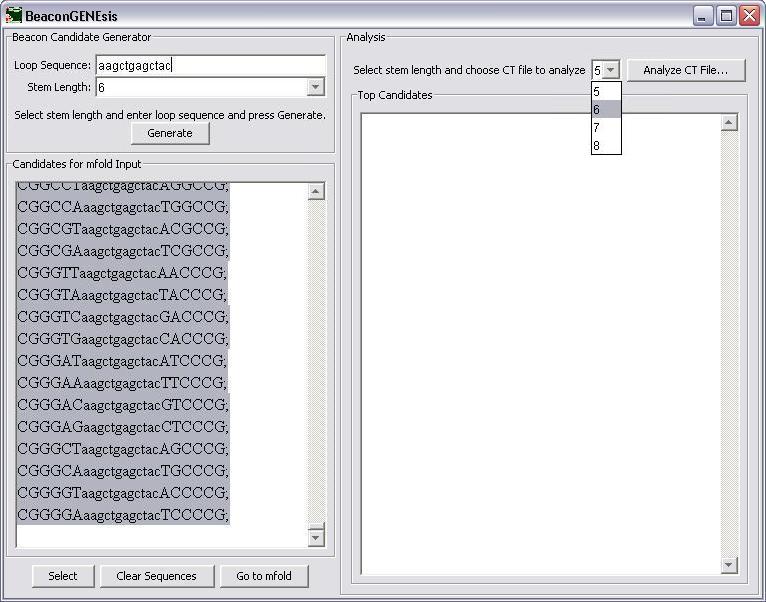
The top 10 candidates are listed ranked by delta G. These can be further analyzed using a self-dimer calculation, like that available from www.idtdna.com